Preface
Working in an HPC environment, the “dependency hell” is the most bothersome nuisance. (Yep, I personally regard it “the most”, not “one of the most”.)
For instance, in R, users often receive messages such as:
"Error: package or namespace load failed for XXX:package YYY was installed before R 4.0.0: please re-install it"
The solution could be relatively easy, you may upgrade to the latest version of R or trying re-installing the YYY package.
In Linux, however, things could be much more painful.
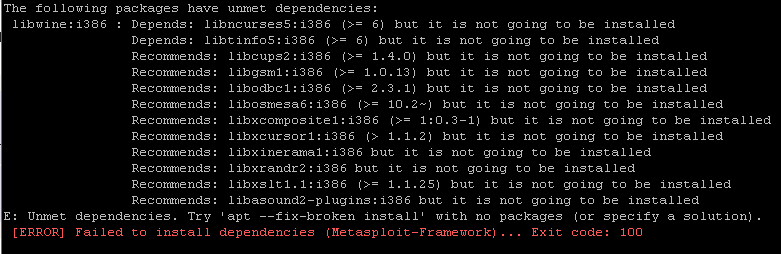
Especially when working in an HPC, it is nearly impossible to install/update pacakges that requires sudo privilege.
Luckily, my ex-boss, Dr. Chang, introduced me the concept of app containerization using tools such as Docker and Singularity. I tried, and realized that I cannot live without them. They are truly my life savers.
In this post, I would like to provide a step by step tutorial on how to run singularity containers in an HPC environment.
Table of Contents
- What is a container?
- Where can I get containers?
2.1 Public resources
2.2 Build my own container - How to run a container on HPC
What is a container?
To begin with, a disclamer: I’m not a CS person. That means I won’t be using terminologies. Instead I’ll use just plain words.
Think of this scenario: you got a message when running a software X on your HPC:
The software X is update, however, the software Y is not. Since software X depends on some functions wrapped in Y, so you are not able to run X now.
– bang, ERROR.
You might think, ok, let me update Y. You tried, and got another message:
You are not allowed to update Y since you are not the super user of this computer.
– bang, ERROR again.
This is the simplest example of dependencies hell. In reality, it could be much more complex since the software dependency manner is not one-to-one; sometimes you may need to install more softwares that requires “sudo” privilege.
One way of solving this is to build your own Utopia, where you take fully control:
- You can keep your softwares at your favourite version, and they don’t auto-update themselves.
- Your softwares has the appropriate dependency softwares, and they always work well with each other.
- Your are the super user of this Utopia, so you can install anything that requires sudo privilege.
- You can still read and write your data (files, images, etc.) to the outside world.
Another advantage of this Utopia is that you can reproduce your analyses – the package version are controlled, so just run your scripts and it’ll always give you the same results – nothing’s gonna change.
This kind of Utopia world is the so called “container”.
As a bioinfo hobbyist and a singularity lover, I’ll be using the bioinformatics related tools to illustrate how to build and use singularity containers. As for the other containerization tools, you can simply check their documentations.
Where can I get containers?
There are two ways of acquiring a container, downloading one from the public, or build one from scratch:
Public resources
Public resources are the easiest way to get a container. Repositories such as Docker Hub and Singularity Hub are the most popular ones. You can search for pre-built containers, such as conda, jupyter notebook, Rstudio, etc. Thesea containers are open source, so you can download them and use them for free.
Build my own container
If you cannot find a container that suits your need, you can build one from scratch. This is a bit more complicated, but it’s not that hard. In this post, I’ll be using the Singularity to illustrate how to build a container.
Basically, what you need is simply a “recipe”. A recipe is a file that contains instructions on how to build a container. Here’s an example of a recipe:
Bootstrap: docker
From: debian:stable-slim
%labels
Version v0.0.1
%help
Please contact Jiayi for help.
%post
## basic stuffs
apt-get update
apt-get install -y --no-install-recommends \
bash-completion \
ca-certificates \
locales
echo 'en_US.UTF-8 UTF-8' >> /etc/locale.gen && \
/usr/sbin/locale-gen en_US.UTF-8 && \
/usr/sbin/update-locale LANG=en_US.UTF-8
## misc stuffs
apt-get update && apt-get install -y --no-install-recommends \
file \
less \
openssl \
libssl-dev \
curl \
coreutils \
gdebi-core
## devel tools
apt-get update && apt-get install -y --no-install-recommends \
g++ \
gfortran \
make \
cmake
## dependencies
apt-get update && apt-get install -y --no-install-recommends \
gettext \
pkg-config \
autoconf-archive \
autoconf \
automake \
autopoint \
txt2man \
build-essential \
autoconf-archive \
automake \
autopoint \
pkg-config \
txt2man
## axel
apt-get -y install aria2
After you’ve done with the recipe, you can paste it into the Singularity Container Builder to generate your own container.
How to run a container on HPC?
In the following code blocks in this section, I’ll add my comments/recommendations starting with ## Jiayi:.
Here, I’ll be using one of my Singularity containers as an example. I’ve built several singularity containers that could boost productivity in bioinformatics research. You can check them from here.
First, in your HPC, you’ll need to have the Singularity installed and loaded. If you don’t have it, you can follow the instructions here. If you have it, you can simply load it:
module load singularity/3.8.3
## Jiayi: check with your HPC admin for the version, or check with `module avail singularity`.
Second, you’ll need to pull the container from the cloud. This above command will pull the container from the cloud and save it as a file named “rserver_4-1-1bioinfo.sif”.
singularity pull --arch amd64 library://jiayiliujiayi/rserver/rserver:4-1-1bioinfo
## Jiayi: This container is a Rstudio server container, along with common R packages including Seurat, ggplot2, tidyverse, dplyr, etc.
Third, you can use this container in your HPC. I’ll use SLURM environment as an example. In your workign directory, you can create a script named “run_rstudio.sh” with the following content:
#!/bin/bash
## Jiayi: This is a SLURM script. You can check with your HPC admin for the details (marked with XXX). You can also check the documentations of SLURM: https://slurm.schedmd.com/
#SBATCH --job-name=rstudio
#SBATCH --output=rstudio_%j.out
#SBATCH --error=rstudio_%j.err
#SBATCH --time=3-00:00:00 ## Jiayi: check with your HPC admin for the time limit, or change it to the time length you need.
#SBATCH --mem=100G
#SBATCH --cpus-per-task=10
#SBATCH --partition=general
#SBATCH --qos=general
#SBATCH --mail-type=ALL
#SBATCH --mail-user=XXX ## Jiayi: change it to your email address if you want to get updated with the job status.
# load the singularity module
module purge
module load singularity/3.8.3
# prepare for the Rstudio server
## Do not suspend idle sessions.
export SINGULARITYENV_RSTUDIO_SESSION_TIMEOUT=0
## Container user set ups
export SINGULARITYENV_USER=XXX ## Jiayi: change it to your username on the HPC
export SINGULARITYENV_PASSWORD=XXXX ## Jiayi: define your own password, and it doesn't necessarily need to be the same as your HPC password.
## Get IP address
IP_ADDR=`hostname -i`
echo $IP_ADDR
## write out the IP address and port number to a file
cat 1>&2 <<END
1. SSH tunnel from your workstation using the following command:
ssh -N -L 8787:${IP_ADDR}:8787 ${SINGULARITYENV_USER}@XXX ## Jiayi: change it to your HPC IP address.
and point your web browser to http://localhost:8787
2. log in to RStudio Server using the following credentials:
user: ${SINGULARITYENV_USER}
password: ${SINGULARITYENV_PASSWORD}
When done using RStudio Server, terminate the job by:
1. Exit the RStudio Session ("power" button in the top right corner of the RStudio window)
2. Issue the following command on the login node:
scancel -f ${SLURM_JOB_ID}
END
# start the Rstudio server
singularity exec --cleanenv \
--bind run:/run,var-lib-rstudio-server:/var/lib/rstudio-server,database.conf:/etc/rstudio/database.conf, $PWD:/XXX/,/tmp \ ## Jiayi: change it to your working directory, and the directory where you want to save the Rstudio session.
~/rserver_4.1.1bioinfo.sif \
--server-user ${USER} \
--auth-none=0 \
--auth-pam-helper-path=pam-helper
printf 'rserver exited' 1>&2
Fourth, you can submit the job to the HPC using the following command:
sbatch run_rstudio.sh
In your working directory, you’ll see two files: “rstudio_XXXX.out” and “rstudio_XXXX.err”. The former one is the output of the job, and the latter one is the error log. You can check the error log if you encounter any problems. If you don’t see any error, you can check the output file. In the output file, you’ll see a line as such:
ssh -N -L 8787:${IP_ADDR}:8787 ${SINGULARITYENV_USER}@XXX
Copy the above command and paste it into your local terminal.
Finally, you can open your web browser and type in “localhost:8787” in the address bar. You’ll see the Rstudio server page. You can log in with your username and password. And Voila. You are in the Rstudio server environment of your Utopia.
Also, you may wanna take a look at the other information in the output file regarding how to terminate the SLURM job.